Presentación de Caso
Hamartoma hipotalámico, a propósito de un caso con evolución inusual
Hypothalamic hamartoma, regarding a case with unusual evolution
Juliet
Pérez González1
https://orcid.org/0009-0006-4117-1318
Aisel Santos
Santos2*
https://orcid.org/0000-0002-3815-2136
1Hospital
General Provincial Universitario "Camilo Cienfuegos". Sancti Spíritus, Cuba.
2Instituto
Nacional de Neurología y Neurocirugía. La Habana, Cuba.
*Autor para la correspondencia. Correo electrónico: aisel.santos@gmail.com
Introducción:
Los hamartomas hipotalámicos son malformaciones originadas en el tuber
cinereume hipotálamo inferior. Tienen un amplio espectro en su presentación
clínica y electroencefalográfica, por lo que su diagnóstico y tratamiento son
todo un desafío.
Objetivo:
Describir la evolución clínica, los resultados de los exámenes complementarios
y la respuesta al tratamiento en una paciente con evolución inusual del hamartoma
hipotalámico.
Caso clínico:
Paciente femenina de 43 años de edad que debuta en la infancia temprana con
crisis gelásticas, sin alteraciones del neurodesarrollo ni endocrinológicas.
En la adolescencia, comienza con crisis de desconexión y tónico-clónicas bilaterales
esporádicas, se diagnostica epilepsia del lóbulo temporal. El aprendizaje, la
conducta y la adaptación social se mantenían normales. Continúa sus estudios
universitarios con excelente rendimiento, a los 36 años se realiza el diagnóstico
de hamartoma hipotalámico. Es actualmente una profesional exitosa, a pesar de
no presentar control con la medicación anticrisis. Recibió una sesión de gamma
knife sin cambios clínicos ni imagenológicos.
Conclusiones:
Se muestra un caso de hamartoma hipotalámico con evolución inusual, presenta
mal control de crisis gelásticas que han evolucionado a status epilépticos,
a pesar de lo cual tiene un desempeño profesional exitoso.
Palabras clave: epilepsias parciales; epilepsia gelástica; hamartoma; neoplasias hipotalámicas.
Introduction:
Hypotalamic hamartomas are malformations originating in the tuber cibereum and
inferior hypothalamus. They have a wide spectrum in their clinical and electroencephalographic
presentation, so their diagnosis and treatment are a challenge.
Objective: Describe the clinical evolution, the results of complementary
examination and the response to treatment in a patient whit unusual evolution
of hypothalamic hamartoma.
Clinical case: 43-year-old female patient who presented in early childhood
with gelastic seizures, without neurodevelopmental or endocrinological problems.
In adolescence, she begins with disconnection seizures and sporadic bilateral
tonic-clonic seizures; temporal lobe epilepsy is diagnosed. Learning, behavior
and social adaptation remained normal. She continued her university studies
with excellent performance, at the age of 36 she was diagnosed with hypothalamic
hamartoma. She is currently a successful professional, despite not being controlled
with anti-seizure medication. She received a gamma knife session without clinical
or imaging changes.
Conclusions: A case of hypothalamic hamartoma with unusual evolution
is shown, she presents poor control of gelastic seizures that have evolved into
status epilepticus, despite which it has a successful professional performance.
Keywords: gelastic epilepsy; hamartoma; hypothalamic neoplasm; partial epilepsies.
Recibido:
11/10/2024
Aceptado:
30/11/2024
INTRODUCCIÓN
Los hamartomas hipotalámicos (HH) son malformaciones originadas en el tuber cinereum e hipotálamo inferior, están compuestos de neuronas ectópicas y tejido glial.(1) Tienen una prevalencia estimada de 1 a 2 casos por 100 000 habitantes y representan una de las causas más notables de epilepsia farmacorresistente.(2)
La presentación clínica clásica es una tríada que incluye crisis gelásticas (CG), retraso en el neurodesarrollo y pubertad precoz central.(2) Sin embargo, los HH tienen un amplio espectro en su presentación clínica,(2,3) que va desde crisis epilépticas esporádicas con cognición normal hasta una encefalopatía epiléptica similar al síndrome de Lennox-Gastaut, con múltiples tipos de crisis epilépticas, trastornos cognitivos y conductuales, en más del 50 % de los pacientes.(4,5)
Para el diagnóstico, debe considerarse la típica presentación clínica de esta enfermedad, así como la evidencia de la lesión por neuroimagen. Sin embargo, esto constituye todo un desafío; muchos casos se diagnostican errónea y tardíamente porque es posible que algunos síntomas no se manifiesten y se piense en otra etiología de la epilepsia.(5)
Se realizó esta investigación con el objetivo de describir la evolución clínica, los resultados de los exámenes complementarios y la respuesta al tratamiento en una paciente con evolución inusual del hamartoma hipotalámico.
CASO CLÍNICO
Paciente femenina de 43 años de edad, con antecedentes prenatales y perinatales normales. Menarquia a los 11 años, menstruaciones regulares. Desempeño escolar, conducta e integración social adecuados. Presentó desde la infancia temprana eventos de carcajadas espontáneas, en la adolescencia continuó con risas inmotivadas más seguidas, precedidas de sensación epigástrica ascendente y rubicundez facial. A los 23 años tuvo 2 crisis tónico-clónicas bilaterales, se diagnosticó una epilepsia focal, se trató con fenitoína y se mantuvo sin control de las crisis.
Alrededor de 2 años después empezó con crisis parciales complejas; se diagnosticó epilepsia del lóbulo temporal, se indicó tratamiento con carbamazepina asociada a valproato y luego a lamotrigina, sin control de crisis.
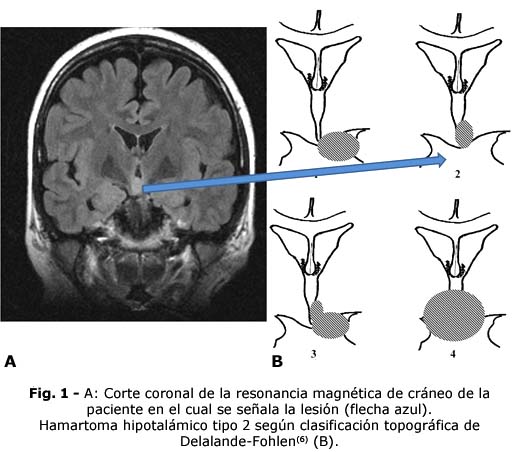
A los 36 años luego de haber cumplido exitosamente misiones internacionalistas y varias responsabilidades laborales, refiere disminución de memoria verbal, de habilidades comunicativas y llanto fácil. Se ingresa en el Instituto de Neurología y Neurocirugía (INN) donde se diagnostica una epilepsia farmacorresistente secundaria a hamartoma hipotalámico (Fig. 1).
Ese año se viabilizó por el Ministerio de Salud Pública el tratamiento con radiocirugía gammaknifeen los EE. UU., se administraron 17 GAY hasta la línea del 50 % isodosis para un volumen 0,3 mL. Se reevaluó cada 3 meses sin evidencias de mejoría clínica ni reducción del hamartoma, según resonancias magnéticas (RM) de cráneo (Fig. 2).
A los 39 años, presentó un status de CG y dacrísticas (llanto), se medicó con fenitoína intravenosa que exacerbó las crisis. Solo se controló con midazolam después de dos días.
Actualmente, continúan las crisis resistentes a fármacos, caracterizadas por una sensación epigástrica ascendente, sabor a sangre, CG que pueden ser leves y en ocasiones prolongadas, asociadas a desconexión y automatismos gestuales simples. En el posictal sabor metálico que puede durar hasta dos días.
Se reingresó en el 2024 en el INN. Al examen físico se evidenció obesidad, deterioro cognitivo leve con alteraciones de la memoria verbal, fluidez verbal fonológica disminuida y disfunción ejecutiva que suple por su alto rendimiento intelectual. Se realizó RM que mostró el hamartoma sin modificaciones. El video electroencefalograma interictal mostró signos de disfunción cortical focal de ligera intensidad y actividad paroxística fronto-temporal bilateral alternante.
La paciente está pendiente de un segundo ciclo de gammaknife, lleva tratamiento con carbamazepina (200 mg) 6 tabletas, levetiracetam (500 mg) 4 tabletas y clonazepam (1 mg) 3 tabletas, diarias. Se trabaja con técnicas de rehabilitación cognitiva, automotivación y autocontrol. Con inserción en distintas actividades del grupo, la evolución es favorable. Ha sufrido incomprensiones por el desconocimiento de su enfermedad en el medio en que se desempeña. Al acudir a urgencias no se reconocen los eventos como crisis epilépticas, sino como trastornos psiquiátricos.
COMENTARIOS
El diagnóstico de los HH representa todo un reto. No existe un factor único que defina la evolución, gravedad y presentación clínica de los pacientes con un HH. se ha observado que existe una relación entre el tamaño, ubicación y tipo de inserción de la lesión.(2,7)
Varios autores plantean(1,2,3) que los hamartomas que afectan la parte posterior del hipotálamo se asocian a crisis epilépticas farmacorresistentes. que generalmente comienzan en la primera infancia y cursan con deterioro cognitivo-conductual, por lo que la edad de inicio de las CG podría tener un papel predictor en la evolución de estos pacientes. En el caso que se presenta, el inicio de las CG desde la infancia con evolución a forma de status epiléptico, no repercutieron en su desarrollo ni desempeño escolar, laboral o integración social.
Por otra parte, las CG pueden simular a las crisis originadas en el lóbulo temporal o frontal, el electroencefalograma no es una prueba específica, muestra gran variabilidad, factores que dificultan más el diagnóstico.(8)
La medicación anticrisis es inefectiva en las CG. La radiocirugía gamma knife es una opción segura y efectiva en el tratamiento del HH, incluso en niños pequeños.(9,10,11)
El conocimiento de estos factores es de gran relevancia para el diagnóstico y manejo integral de los pacientes.
Se muestra un caso clínico de hamartoma hipotalámico con evolución inusual, que según Islas-García D y otros(2) muchos neurólogos pueden encontrarse con sólo 1 o 2 de estos casos en su trayectoria, que a pesar de la epilepsia farmacorresistente y la mala respuesta a todas las terapias utilizadas se mantiene a los 43 años con un desempeño profesional exitoso.
Ética y consentimiento
Los autores presentan el consentimiento informado del paciente para la publicación del trabajo.
REFERENCIAS BIBLIOGRÁFICAS
1. Conde Blanco E, Anciones Martín C, Manzanares I, Gil López F, Roldán P, Donaire A, et al. Hypothalamic hamartomas in adulthood: clinical spectrum and treatment outcome-A unicenter experience [Internet]. Brain Behav. 2019; 9(11):e01412. DOI: 10.1002/brb3.1412
2. Islas-García D, Alderete-Berzaba J, Queiros-Serna CV, Perera-Canul RN. Crisis gelásticas secundarias a hamartoma hipotalámico [Internet]. An Med Asoc Med Hosp ABC. 2020; 65(3):233-8. DOI: 10.35366/95681.
3. Striano S, Striano P. Clinical features and evolution of the gelastic seizures-hypothalamic hamartoma syndrome [Internet]. Epilepsia. 2017; 58(2):12-5. DOI: 10.1111/epi.13753
4. Cross H, Spoudeas H. Medical management and antiepileptic drugs in hypothalamic hamartoma. Epilepsia [Internet]. 2017; 58:16-21. DOI: 10.1111/epi.13758
5. Wilfong A, Curry D. Hypothalamic hamartomas: optimal approach to clinical evaluation and diagnosis. Epilepsia [Internet]. 2013; 54(9):109-14. DOI:10.1111/epi.12454
6. Delalande O, Fohlen M. Disconnecting surgical treatment of hypothalamic hamartoma in children and adults with refractory epilepsy and proposal of a new classification [Internet]. Neurol Med Chir (Tokyo). 2003; 43(2):61-8. DOI: 10.2176/nmc.43.61. PMID: 12627881.
7. Harrison V, Oatman O, Kerrigan J. Hypothalamic hamartoma with epilepsy: Review of endocrine comorbidity [Internet]. Epilepsia. 2017; 58(2):50-9. DOI: 10.1111/epi.13756
8. Troester M, Haine-Schlagel R, Ng Y, Chapman K, Chung S, Drees C, Kerrigan J. EEG and video-EEG seizure monitoring has limited utility in patients with hypothalamic hamartoma and epilepsy [Internet]. Epilepsia. 2011; 52(6):1137-43. DOI: 10.1111/j.1528-1167.2011.03095.x
9. Ferrand-Sorbets S, Fohlen M, Delalande O, Zuber K, Bulteau C, Levy M, Chamard P, Taussig D, Dorison N, Bekaert O, Tisdall M, Chipaux M, Dorfmüller G. Seizure outcome and prognostic factors for surgical management of hypothalamic hamartomas in children [Internet]. Seizure. 2020; 75:28-33. DOI: 10.1016/j.seizure. 2019.11.013.
10. Bourdillon P, Ferrand-Sorbet S, Apra C, Chipaux M, Raffo E, Rosenberg S, Bulteau C, Dorison N, Bekaert O, Dinkelacker V, Le Guérinel C, Fohlen M, Dorfmüller G. Surgical treatment of hypothalamic hamartomas [Internet]. Neurosurg Rev. 2021; 44(2):753-62. DOI: 10.1007/s.10143-020-01298-z
11. Régis J, Lagmari M, Carron R, Hayashi M, McGonigal A, Daquin G, Villeneuve N, Laguitton V, Bartolomei F, Chauvel P. Safety and efficacy of Gamma Knife radiosurgery in hypothalamic hamartomas with severe epilepsies: A prospective trial in 48 patients and review of the literature [Internet]. Epilepsia. 2017; 58(Suppl 2):60-71. DOI: 10.1111/epi.13754
Conflictos de interés
Los autores declaran que no existen conflictos de interés.
Información financiera
No se declaran fuentes de financiamiento.
Disponibilidad de datos
Los datos utilizados para la presentación del caso se encuentran en la historia clínica de la paciente archivada en el Instituto de Neurología y Neurocirugía.
{kind=link}