Presentación de Caso
Encefalitis de Rasmussen de debut tardío en una adolescente de 19 años
Late-onset Rasmussen's encephalitis in a 19-year-old adolescent
Leidy García Morales1* https://orcid.org/0000-0003-2627-4790
Juan Carlos Padilla Reyes1 https://orcid.org/0009-0001-2111-8441
Aisel Santos Santos1 https://orcid.org/0000-0002-3815-2136
Mercedes Rita Salinas Olivares2 https://orcid.org/0000-0003-0909-5133
Karenia Joglar Hernández3 https://orcid.org/0000-0002-1793-4390
1Instituto de Neurología y Neurocirugía. Servicio Neurología. La Habana, Cuba.
2Instituto de Neurología y Neurocirugía. Departamento de Anatomía Patológica. La Habana, Cuba.
3Instituto de Neurología y Neurocirugía. Departamento de Imagenología. La Habana, Cuba.
*Autor para la correspondencia. Correo electrónico: leidy.gmc@gmail.com
RESUMEN
Introducción: La encefalitis de Rasmussen es una enfermedad neurológica progresiva poco frecuente y de etiología aún desconocida. Se caracteriza por crisis epilépticas polimórficas, frecuentes y de difícil control, deterioro neurocognitivo y evidencia radiológica de hemiatrofia cerebral, de presentación en la infancia y de pronóstico desfavorable.
Objetivo: Documentar la historia clínica de una paciente con inicio y evolución atípicos de la Encefalitis de Rasmussen.
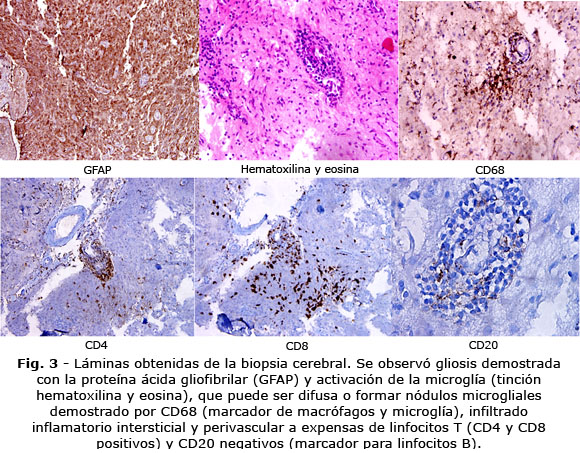
Caso Clínico: Fémina de 19 años de edad que presentó cambios en su conducta social. Tres meses después comienza con hemiparesia izquierda progresiva y crisis focales disperceptivas con síntomas motores, algunas podían evolucionar a tónico clónica bilateral. En la resonancia magnética de cráneo se observó hemiatrofia cerebral progresiva. Se realiza biopsia cerebral, con intensa gliosis a predominio de astrocitos gemastocíticos, presencia escasa de neuronas retraídas con núcleos picnóticos, evidencia de daño neuronal crónico grave, abundantes microglías e inflamación perivascular de linfocitos T diagnosticándose una encefalitis de Rasmussen.
Conclusiones: La encefalitis de Rasmussen es una enfermedad infrecuente pero devastadora. Su diagnóstico y tratamiento precoces son claves en la evolución de los pacientes y aumentan las posibilidades de reinserción en la vida social.
Palabras clave: crisis epilépticas; encefalitis; síndrome de Rasmussen.
ABSTRACT
Introduction: Rasmussen's encephalitis is a rare progressive neurological disease of unknown etiology. It is characterized by frequent, difficult-to-control polymorphic epileptic seizures, neurocognitive impairment, and radiological evidence of cerebral hemiatrophy, with onset in childhood and poor prognosis.
Objective: To document the clinical history of a patient with atypical onset and progression of Rasmussen's encephalitis.
Clinical case: A 19-year-old female, whose initial manifestation was behavioral disorder. Three months later, she presented progressive left hemiparesis and focal disperceptive seizures with motor symptoms, some of which could evolve into bilateral tonic clonic. Progressive cerebral hemiatrophy was observed on cranial magnetic resonance imaging. A brain biopsy was performed, which showed severe gliosis with predominance of gemastocytic astrocytes, scarce presence of retracted neurons with pyknotic nuclei, evidence of severe chronic neuronal damage, abundant microglia and perivascular inflammation of T lymphocytes, diagnosing Rasmussen's encephalitis.
Conclusions: Rasmussen's encephalitis is a rare but devastating disease. Early diagnosis and treatment are key in the evolution of patients with and increase the chances of reintegration into social life.
Keywords: encephalitis; epileptic seizures; Rasmussen syndrome.
Recibido: 14/01/2025
Aprobado: 04/08/2025
INTRODUCCIÓN
La encefalitis de Rasmussen (ER) fue descrita por primera vez en 1958 por Theodore Rasmussen como una encefalitis hemisférica crónica que causaba atrofia unilateral. Es un raro trastorno neurológico crónico, caracterizado por epilepsia farmacorresistente y deterioro neurológico y cognitivo progresivo debido a una inflamación unilateral de la corteza cerebral.(1) La incidencia anual es de unos 2,4 casos/107 personas menores de 18 años, con edad media de presentación entre 6 y 8 años.(2) Inicia en muchos casos con crisis epilépticas focales; en las que la epilepsia parcial continua constituye su presentación hasta en un 50 % de los casos.(3) De forma excepcional, el inicio de las crisis está precedido por hemiparesia, hemidistonía o hemiatetosis de progresión lenta.(4,5) Durante la fase temprana de la enfermedad las crisis epilépticas pueden ser escasas y el electroencefalograma (EEG) y la resonancia magnética de cráneo (RM) suelen ser normales.(3) Como resultado, el diagnóstico precoz es difícil.
El objetivo propuesto para esta presentación es documentar la historia clínica de una paciente con inicio y evolución atípicos de la Encefalitis de Rasmussen.
CASO CLÍNICO
Paciente femenina de 19 años de edad con antecedentes aparentes de salud. Se presentó en el Instituto de Neurología y Neurocirugía con 6 meses de evolución de crisis epilépticas focales disperceptivas motoras: Desconexión del medio – elevación de miembro superior izquierdo – postura tónica asimétrica de varios minutos de duración. Además, de un status epiléptico convulsivo con manejo en sala de terapia intensiva de su provincia. Las crisis epilépticas se hicieron frecuentes. Después de esto, se notó dificultad en la marcha que se instaló de manera progresiva y debilidad del miembro superior izquierdo. La familia refirió que a las crisis epilépticas le antecedió cambios en la conducta, con tendencia a la agresividad e irritabilidad. A la llegada al Instituto había abandonado las actividades académicas.
Examen físico neurológico:
Hemiparesia espástica izquierda.
Hiperreflexia bilateral con áreas reflexógenas aumentadas, reflejos policinéticos, respuesta clonoide (predominio izquierdo).
Hipoestesia, hipoalgesia en hemicuerpo izquierdo.
Hoffman y Babinski bilaterales (izquierdo espontáneo).
Clonus patelar y de tobillo bilaterales.
Evaluación neuropsicológica: Memoria episódica alterada. Lenguaje sin alteraciones. Baja fluidez semántica y fonemática. Funciones audio motrices perseverantes, impulsivas y con falta de secuencia. Alteración del procesamiento espacial.
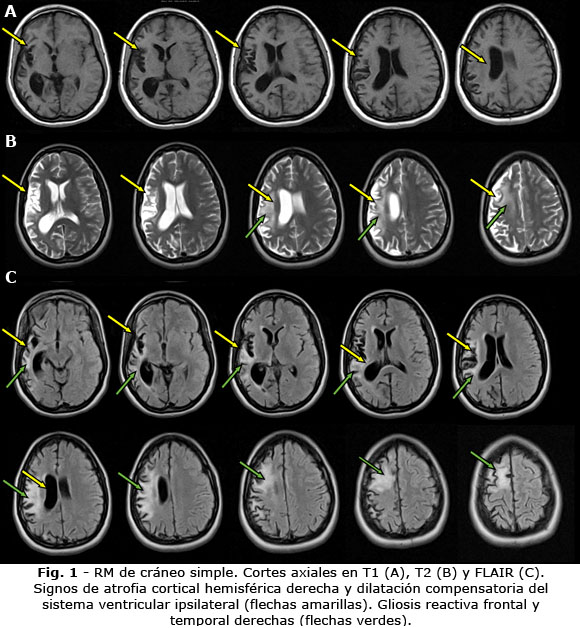
Como parte del protocolo de diagnóstico se realizó Resonancia Magnética de cráneo de alto campo (Fig. 1), EEG (Fig. 2) y biopsia cerebral (Fig. 3).
Tratamiento y evolución: Se diagnosticó ER. Se reajustó su tratamiento anticrisis y optimizó la carbamazepina hasta 600 mg/día. Se administraron 5 ciclos de Intacglobin (0,4 gramos/kg/día/5 días), con un intervalo de 30 días cada ciclo. En las consultas de seguimiento se evidenció control de las crisis epilépticas y detención de la progresión de la hemiparesia y el deterioro cognitivo. Un año después del diagnóstico la paciente comenzó su primer embarazo, que concluyó sin complicaciones. Hasta la fecha se mantiene estable de su epilepsia.
COMENTARIOS
La ER es una entidad poco frecuente, pero con alta índice de discapacidad. Durante su fase aguda las crisis epilépticas se vuelven frecuentes, aparece deterioro cognitivo progresivo, hemiparesia y hemianopsia.(3) En casos con afectación del hemisferio dominante el lenguaje puede verse afectado, con afasia motora.(6) En la fase residual las crisis epilépticas pueden volverse menos frecuentes con déficits neurológicos permanentes.(6,7)
Para su diagnóstico deben estar presentes los 3 criterios de la Parte A o 2 de los 3 de la Parte B. Si no es posible realizar biopsia, es necesario RM con gadolinio y Tomografía de cráneo para documentar la ausencia de realce con el contraste y calcificaciones como principales diferenciales de una vasculitis unihemisférica.
Parte A:
1) Crisis focales (con o sin Epilepsia parcial continua) y déficit(s) cortical(es) unilateral(es)
2) Enlentecimiento unihemisférico con o sin actividad epileptiforme y comienzo ictal unilateral
3) Atrofia cortical focal unihemisférica y al menos una de las siguientes alteraciones:
Hiperintensidad de la sustancia gris o blanca en T2/FLAIR
Hiperintensidad o atrofia de la cabeza del caudado ipsilateral
Parte B:
1) Epilepsia parcial continua o déficit cortical unilateral progresivo
2) Atrofia cortical focal unihemisférica progresiva
3) Histopatología: Encefalitis con infiltrado de células T y células microgliales activadas y astrogliosis reactiva. Numerosos macrófagos parenquimatosos. La presencia de células plasmáticas, células B o cuerpos de inclusión virales excluyen el diagnóstico de ER.(8)
La etiología del ER aún no se dilucida, aunque se identifica como un proceso inflamatorio crónico. Las hipótesis etiopatogénicas con mayor consenso son la infección por virus neurotropos o la posibilidad de un proceso autoinmune al demostrarse en algunos pacientes la existencia de anticuerpos contra el receptor 3 del glutamato cerebral (anti -GluR3) o anticuerpos anti-descarboxilasa del ácido glutámico (anti-GAD), o el aumento de la mediación celular T en el proceso inflamatorio cerebral.(3,9,10) Se desconoce el motivo de la afectación unilateral exclusiva.
Hasta el momento ningún fármaco concibe la capacidad de lograr un control completo de la progresión de la enfermedad. La cirugía representa la única opción definitiva para controlar las convulsiones y detener el deterioro neurológico en pacientes con ER.(2,3,11) Menos del 10 % de los pacientes con ER debutan después de los 18 años.(12)
Se presenta un caso clínico de ER con atipicidades en su debut y progresión (debut a los 19 años con manifestaciones conductuales, escasa frecuencia de crisis y remisión de la enfermedad con tratamiento farmacológico). Hasta el momento la paciente se encuentra con actividad social y económica. El diagnóstico y tratamiento oportunos lograron controlar una enfermedad que con frecuencia es progresiva, de gran gravedad y discapacitante.
Se concluye que, la ER es una enfermedad infrecuente pero devastadora. Su diagnóstico y tratamiento precoces son claves en la evolución de los pacientes y aumentan las posibilidades de reinserción en la vida social.
Ética y consentimiento
Se obtuvo consentimiento de la paciente y la familia para la publicación del caso.
REFERENCIAS BIBLIOGRÁFICAS
1. Cay-Martinez KC, Hickman RA, McKhann Ii GM, Provenzano FA, Sands TT. Rasmussen Encephalitis: An Update [Internet]. Semin Neurol. 2020; 40(2):201-10. DOI: 10.1055/s-0040-1708504
2. Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M, et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins [Internet] Epilepsia. 2013; 54(3):543-50. DOI: 10.1111/epi.12042
3. Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, et al. Rasmussen's encephalitis: clinical features, pathobiology, and treatment advances [Internet] Lancet Neurol. 2014 ;13(2):195-205. DOI: 10.1016/S1474-4422(13)70260-6
4. Bhatjiwale MG, Polkey C, Cox TC, Dean A, Deasy N. Rasmussen's encephalitis: neuroimaging findings in 21 patients with a closer look at the basal ganglia [Internet]. Pediatr Neurosurg. 1998; 29(3):142-8. DOI: 10.1159/000028709
5. Bien CG, Elger CE, Leitner Y, Gomori M, Ran B, Urbach H, et al. Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed-onset seizures [Internet]. Eur J Neurol. 2007; 14(4):387-90. DOI: 10.1111/j.1468-1331.2007.01684.x
6. Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD, et al. The natural history of Rasmussen's encephalitis [Internet]. Brain. 2002;125(Pt 8):1751-9. DOI: 10.1093/brain/awf176
7. Garófalo-Gómez N, Hamad AP, Centeno RS, Ferrari TP, Carrete Jr. H, Caboclo LO, et al. Evolución posquirúrgica en pacientes con encefalitis de Rasmussen operados por hemisferotomía [Internet]. Rev Neurol. 2013; 56(04):214-19 DOI: 10.33588/rn.5604.2012419
8. Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement [Internet]. Brain. 2005; 128(Pt 3):454-71. DOI: 10.1093/brain/awh415
9. Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies [Internet]. Epilepsia. 2004; 45(5):516-26. DOI: 10.1111/j.0013-9580.2004.33103.x
10. Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis [Internet]. Science. 1994, 29:265(5172):648-51. DOI: 10.1126/science.8036512
11. Hoffman CE, Ochi A, Snead OC 3rd, Widjaja E, Hawkins C, Tisdal M, et al. Rasmussen's encephalitis: advances in management and patient outcomes [Internet]. Childs Nerv Syst. 2016; 32(4):629-40. DOI: 10.1007/s00381-015-2994-x
12. Dupont S, Gales A, Sammey S, Vidailhet M, Lambrecq V. Late-onset Rasmussen Encephalitis: A literature appraisal [Internet]. Autoimmun Rev. 2017; 16(8):803-10. DOI: 10.1016/j.autrev.2017.05.022
Conflictos de interés
Los autores declaran que no exististe conflicto de interés.
Información financiera
Los recursos utilizados para el estudio y tratamiento de este paciente fueron suministrados al Instituto por el Ministerio de Salud Pública de la República de Cuba.
Disponibilidad de datos
Los datos utilizados para la presentación del caso corresponden al Instituto de Neurología y Neurocirugía de Cuba.